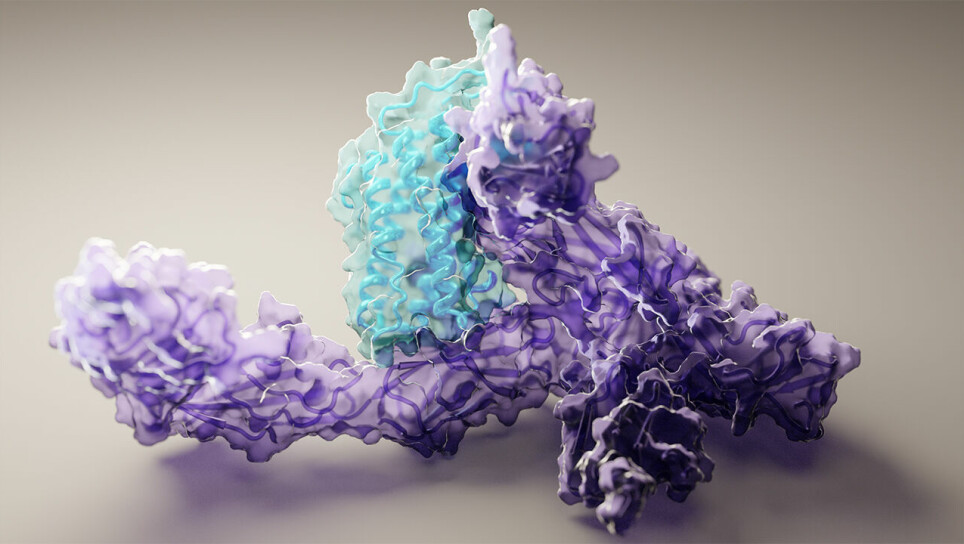
I nesten 50 år har forskere strevd med å løse en av naturens mest forvirrende utfordringer: proteinfolding.
Men i år har det skjedd et gjennombrudd: Forskere har vist at AI-drevet programvare kan forutsi tusenvis av nøyaktige proteinstrukturer. Tidsskriftet Science har kåret det til årets gjennombrudd.
To prosjekter får æren: AlphaFold og RoseTTA-fold.
Ole Winther, professor i datavitenskap ved DTU og genomisk bioinformatikk ved Københavns Universitet, forstår godt kåringen. Fordi det er «et veldig, veldig vanskelig problem». Og forskere har brukt mange år – ja, rundt 50 – på å finne en måte å forutsi de tredimensjonale strukturene til proteiner.
– Dette er et virkelig gjennombrudd. Det har vært mye hype om AI, maskinlæring og dyplæring de siste 5 til 10 årene. Men dette er første gang AI har gitt et så stort bidrag til et vitenskapelig gjennombrudd, sier Winther.
Den entusiasmen deler Daniel Otzen, som forsker på proteinfolding, feilfolding og aggregering ved iNANO ved Aarhus Universitet.
– Det er fullt berettiget. AlphaFold og RoseTTA-fold har lært å bruke de kjente strukturene – opptil 100.000 – for å kunne finne mønstre og forutsi nesten alle andre proteiner. De har bygget på en enorm mengde eksperimentelt arbeid fra de siste 50 årene, sier Otzen, som også har skrevet en forskningsartikkel om AlphaFold og RoseTTA-fold i tidsskriftet J. Mol. Biol.
Proteiner er utrolig viktige
Proteiner er cellenes arbeidshester; de flytter oksygen, molekyler til å utføre alle mulige oppgaver, sørger for at det skjer kjemiske reaksjoner slik at vi kan omdanne mat til energi. I tillegg bygger de nye materialer i cellene og hjelper cellene til å formere seg. Det forklarer Daniel Otzen.
– De gjør alt man kan tenke seg og mer til. Grunnen til dette er at de fleste har en bestemt tredimensjonal struktur. De store kodene er sammensatt av 20 forskjellige aminosyrer som har ulike fysisk-kjemiske egenskaper, sier han.
– Trikset er at proteiner består av sekvenser av aminosyrerester satt sammen i en bestemt rekkefølge. Bare 20 naturlige aminosyrer er satt sammen i mange kombinasjoner – i en nesten uendelighet av mulige proteinsekvenser.
Om proteiner
- Proteiner er viktige bestanddeler av alle levende organismer. De hjelper til med å bygge alt fra organer til hud og utfører en rekke oppgaver inne i cellene.
- Antistoffer er også en type protein.
- Forskere har identifisert rundt 200 millioner proteiner, men de kjenner bare formen til en liten del av dem.
- Ved å se på gener som koder for proteiner, kan forskere bestemme rekkefølgen på aminosyrene de er laget av.
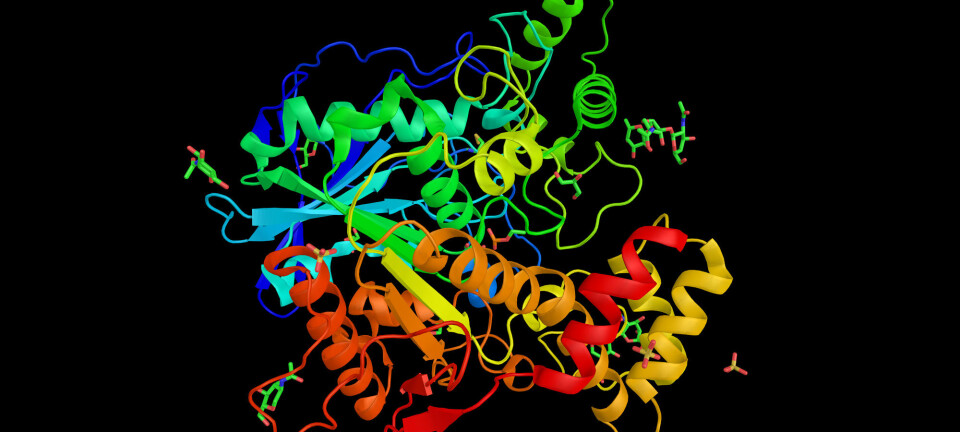
- Men proteiner er ikke bare tråder. De kan også krølle seg og brettes til intrikate strukturer. Det er først når proteinene har funnet sin form at egenskapene deres kommer frem.
- I teorien kan proteinet foldes på astronomisk mange forskjellige måter. Men hva som styrer hvordan proteinet tar form, er ikke helt kjent. Men kunstig intelligens har evnen til å se mønstre som hittil har vært skjult for oss.
Den nøyaktige sammensetningen av rekkefølgen avgjør om og hvordan proteinene folder seg som en bestemt struktur.
– Strukturen er det vi kaller aktivitetsgrunnlaget. Struktur gir funksjon. Forskere har jobbet med dette siden det ble klart at proteiner hadde visse strukturer, sier Otzen.
En av biologiens hellige graler
Proteinfolding handler om å kunne forutsi den tredimensjonale strukturen til et protein fra en enkel aminosyresekvens. Det er en av biologiens mange hellige graler, ifølge Ole Winther.
– Formen og strukturen til proteiner er avgjørende for de fleste prosessene i menneskekroppen, men proteinstrukturer er ikke bare avgjørende for å forstå biologiske fenomener, forklarer han.
– Vi kan bruke proteinfolding for å forstå biologi. Biologi handler om hvordan proteiner fungerer. De har en struktur, visse deler sitter på overflaten og samhandler med andre proteiner, molekyler og DNA. Du kan man nå forutsi. Så det gir oss bedre verktøy for å designe og forstå biologiske prosesser, forklarer Winther.
Gjennombrudd på to fronter
Ifølge Science er det et gjennombrudd på to fronter:
For det første løser det et 50 år gammelt vitenskapelig problem. For det andre er det en innovativ teknikk som vil fremskynde vitenskapelig oppdagelse.
«Det er et enormt gjennombrudd og åpner for mye fremtidig forskning», skriver sjefredaktøren for Science, H. Holden Thorp, i en lederartikkel.
En gang i tiden var det en tidkrevende og kostbar prosess å bestemme strukturen til et protein i laboratoriet.
Datamodeller for proteinfolding har vært under utvikling i flere tiår, men det er først lykkes med AlphaFold og Rosetta, som ble presentert med artikler i Nature og Science.
DeepMind revolusjonerte CASP-konkurransen
Begge prosjektene har deltatt i CASP-konkurransen. CASP står for Critical Assessment of Structure Prediction. Annethvert år siden 1994 har den tatt temperaturen på forskernes evne til å folde proteiner via en datamaskin. Det har gått fremover, men det var først i 2018 det virkelig skjedde noe som fikk oppmerksomhet.
I 2018 deltok DeepMind, eid av Google, for første gang i konkurransen med sitt AlphaFold-system. Det ble sett på som et stort fremskritt, mener Ole Winther.
– I 2020 kom AlphaFold2, som etter CASPs standarder kan sies å ha løst problemet med proteinfolding, forklarer Winther.
Det kan på sikt brukes til å designe molekyler og dermed bidra til å revolusjonere medisinen.
Effekten kan være større enn antatt
DeepMinds metode kan imidlertid vise seg å ha enda større effekt på måten man forsker på, og spørsmålet er om forskningsverdenen er klar til å utløse det potensialet. Winther skrev en artikkel om dette i danske Science News i mars 2021.
– Nå kan en datamaskin regne ut hvordan proteiner ser ut så lenge du har sekvensen. Rekkefølgen er lett å finne. Hver gang du sekvenserer for eksempel et koronavirus, finner du sekvensen til proteinene, forklarer Daniel Otzen.
– DeepMind har utnyttet Googles enorme datakraft og kombinere med smarte nettverksmetoder for å analysere strukturer. Plutselig kunne de mer nøyaktig hvordan proteinstrukturer så ut.
Minst like imponert over RoseTTA
RoseTTA-fold deltok også i konkurransen. De var veldig gode på et tidlig stadium, men de holdt seg til visse metoder: De tok fragmenter av små biter av protein og satte dem sammen i sin spådom, forklarer Ole Winther.
DeepMind var mye bedre, men publiserte bare metoden i grove trekk. RoseTTA ble opptatt med å finne ut hva DeepMind gjorde. Det var et kappløp med tiden, forklarer Winther.
I samme uke som DeepMind beskrev metoden deres i tidsskriftet Nature, fikk RoseTTA sin artikkel i Science.
Daniel Otzen er imidlertid også ganske imponert over RoseTTA.
– Dette er to konkurrenter som bruker samme teknikk. AlphaFold fortjener honnør for å være den første. Men RoseTTA har klart det uten tilgang til massiv datakraft. Så jeg er minst like importert over RoseTTA, understreker han.
En ny verden
Vi går inn i en «ny verden av medisin» sier Ole Winther.
– Legemiddelselskapene har alltid sett manuelt se på hvordan visse potensielle medikamenter kommer inn i proteiner. Dette blir nå enklere.
Det er mange prosesser vi kan automatisere, sier Winther.
– Det vil endre mye på hvordan man lager medisiner og hvordan man lager materialer. Alle disse AI-metodene vil virkelig endre mye rundt vitenskap og teknologi, legger han til.
Daniel Otzen er enig.
– Vi har kommet litt nærmere. Kunnskapen vår om vår verden rundt oss har flyttet seg. Det vil gjennomsyre vår innsikt og kunnskap i tiden som kommer, sier han.
Ny DNA-teknologi, MDMA mot PTSD og covid-19-piller
Hvert år markerer det anerkjente tidsskriftet Science årets største vitenskapelige gjennombrudd.
De markerer også en rekke av de nest største gjennombruddene .
- Ny teknologi gjør det mulig for forskere å «kjemme» tre huler med 25.000 år gammel jord for DNA fra mennesker og dyr.
- Forskere har kommet et skritt nærmere å kunne produsere fusjonsenergi, og det kan være en ny start for energiformen som krever enorme mengder varme.
- Nye piller kan redusere risikoen for alvorlig sykdom på grunn av COVID-19.
- Ny forskning tyder på at det psykedeliske stoffet MDMA kan redusere symptomene hos pasienter med PTSD.
- Kunstige antistoffer produsert i et laboratorium kan temme infeksjonssykdommer som COVID-19 og til slutt bli et våpen mot fremtidige pandemier.
- Nye data fra NASA gir en forståelse av jordskjelv på Mars.
- Forskere lærer mer om elementærpartikler, og det kan endre standardmodellen for fysikk. Forskere fra det amerikanske Fermi National Accelerator Laboratory har sporet opp den ukjente partikkelen ved å studere en annen partikkel, som kalles en myon.
- Genteknologi tas til et nytt nivå. For første gang har CRISPR-teknologi blitt injisert direkte i pasienter og forbedret synet hos pasienter som har arvelige synsproblemer.
- For første gang har forskere klart å dyrke musefoster i mer enn 3 til 4 dager – utenfor morens kropp. Det kan bidra til forståelsen av de tidlige stadiene av menneskelig utvikling.
Referanser:
H. Holden Thorp: Proteins, proteins everywhere. Science (lederartikkel), 2021. DOI: 10.1126 / science.abn5795
John Jumper mfl.: Highly accurate protein structure prediction with AlphaFold. Nature, 2021. DOI: 10.1038/s41586-021-03819-2
Minkyung Baek mfl.: Accurate prediction of protein structures and interactions using a three-track neural network. Science, 2021. (Sammendrag) DOI: 10.1126/science.abj8754
© Videnskab.dk. Oversatt av Lars Nygaard for forskning.no. Les originalsaken på videnskab.dk her.
Vi vil gjerne høre fra deg!
TA KONTAKT HER
Har du en tilbakemelding, spørsmål, ros eller kritikk? Eller tips om noe vi bør skrive om?